Code Walkthrough: A use case of YY1 and YY2 in HeLa cells
Mahmoud Ahmed1
2020-12-16
Source:vignettes/workshop_code.Rmd
workshop_code.Rmd
# load required libraries
library(dplyr)
library(tidyr)
library(readr)
library(purrr)
library(GenomicRanges)
library(rtracklayer)
library(AnnotationDbi)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
library(org.Hs.eg.db)
library(target)Motivation
YY1 and YY2 belongs to the same family of transcription factors.
-
Ying Yang 1 (YY1)
- A zinc finger protein
- Direct deacetylase and histone acetyltransferases of many promoters
- Induces or represses the expression of the target genes
-
Ying Yang 2 (YY2)
- A zinc finger protein
- Arose by retro-transposition of YY1
Using the target analysis, we will attempt to answer the following questions:
- Do the two transcription factors share the same target genes?
- What are the consequences of the binding of each factor on its targets?
- On the shared targets, how the two factors functionally interact?
Datasets
To answer these questions, we use publicly available datasets.
Table 1. Expression and binding data of YY1 and YY2 in HeLa cells.
| GEO ID | Data Type | Design | Ref. |
|---|---|---|---|
| GSE14964 | Microarrays | YY#-knockdown | Chen et al. (2010) |
| GSE31417 | ChIP-Seq | YY1 vs input | Michaud et al. (2013) |
| GSE96878 | ChIP-Seq | YY2 vs input | Wu et al. (2017) |
Data pre-processing:
- Microarrays were obtained in the form of differential expression between the two conditions from KnockTF.
- The ChIP peaks were obtained in the form of
bedfiles from ChIP-Atlas. - USSC hg19 human genome to extract the genomic annotation
Data analysis:
- Prepare the three sources of data for the target analysis
- Predict the specific targets for each individual factors
- Predict the combined function of the two factors on the shared target genes
Preparing the binding data
The ChIP peaks were downloaded in the form of separate bed files for each factor. We first locate the files in the data/ directory and load the files using import.bed. Then the data is transformed into a suitable format, GRanges. The resulting object, peaks, is a list of two GRanges items, one for each factor.
# locate the peaks bed files
peak_files <- c(YY1 = 'data/Oth.Utr.05.YY1.AllCell.bed',
YY2 = 'data/Oth.Utr.05.YY2.AllCell.bed')
# load the peaks bed files as GRanges
peaks <- map(peak_files, ~GRanges(import.bed(.x)))
# show the numbers of peaks of the two factors
lengths(peaks)
#> YY1 YY2
#> 31490 3368
# show the first few entries of the GRanges object
show(peaks$YY1)
#> GRanges object with 31490 ranges and 4 metadata columns:
#> seqnames ranges strand | name score
#> <Rle> <IRanges> <Rle> | <character> <numeric>
#> [1] chr1 9980-10149 * | ID=SRX092576;Name=YY.. 186
#> [2] chr1 10001-10265 * | ID=SRX190209;Name=YY.. 512
#> [3] chr1 10368-10452 * | ID=SRX190209;Name=YY.. 128
#> [4] chr1 11219-11322 * | ID=SRX190209;Name=YY.. 100
#> [5] chr1 29140-29432 * | ID=SRX190209;Name=YY.. 418
#> ... ... ... ... . ... ...
#> [31486] chrY 58996509-58996690 * | ID=SRX092576;Name=YY.. 135
#> [31487] chrY 58996927-58997108 * | ID=SRX092576;Name=YY.. 183
#> [31488] chrY 58996934-58997092 * | ID=SRX190209;Name=YY.. 111
#> [31489] chrY 59363025-59363426 * | ID=SRX190209;Name=YY.. 894
#> [31490] chrY 59363027-59363397 * | ID=SRX092576;Name=YY.. 404
#> itemRgb thick
#> <character> <IRanges>
#> [1] #00BDFF 9980-10149
#> [2] #0CFF00 10001-10265
#> [3] #0082FF 10368-10452
#> [4] #0066FF 11219-11322
#> [5] #00FF53 29140-29432
#> ... ... ...
#> [31486] #0089FF 58996509-58996690
#> [31487] #00BAFF 58996927-58997108
#> [31488] #0071FF 58996934-58997092
#> [31489] #FF6C00 59363025-59363426
#> [31490] #00FF61 59363027-59363397
#> -------
#> seqinfo: 56 sequences from an unspecified genome; no seqlengths
# show number of peaks in each chromosome
table(seqnames(peaks$YY1))
#>
#> chr1 chr10 chr11
#> 3126 1632 1590
#> chr12 chr13 chr14
#> 1817 605 971
#> chr15 chr16 chr17
#> 964 1246 1613
#> chr17_gl000204_random chr17_gl000205_random chr18
#> 1 12 548
#> chr19 chr19_gl000208_random chr1_gl000191_random
#> 1626 18 4
#> chr1_gl000192_random chr2 chr20
#> 4 2304 775
#> chr21 chr22 chr3
#> 332 728 1935
#> chr4 chr4_gl000193_random chr4_gl000194_random
#> 1369 1 1
#> chr5 chr6 chr7
#> 1479 1688 1538
#> chr7_gl000195_random chr8 chr9
#> 11 1134 1330
#> chr9_gl000198_random chr9_gl000199_random chrM
#> 1 37 10
#> chrUn_gl000211 chrUn_gl000212 chrUn_gl000214
#> 3 4 9
#> chrUn_gl000216 chrUn_gl000218 chrUn_gl000219
#> 44 1 5
#> chrUn_gl000220 chrUn_gl000221 chrUn_gl000222
#> 37 1 5
#> chrUn_gl000223 chrUn_gl000224 chrUn_gl000225
#> 4 8 51
#> chrUn_gl000226 chrUn_gl000227 chrUn_gl000228
#> 18 1 12
#> chrUn_gl000235 chrUn_gl000237 chrUn_gl000240
#> 3 1 1
#> chrUn_gl000242 chrUn_gl000243 chrUn_gl000245
#> 1 1 1
#> chrX chrY
#> 630 199
# show the the width of the peaks
summary(width(peaks$YY1))
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 85.0 111.0 151.0 192.9 230.0 1772.0
# show the strands of the peaks
unique(strand(peaks$YY1))
#> [1] *
#> Levels: + - *Preparing the expression data
The differential expression data were downloaded in tabular format.
- Locate the files in
data/ - Read the files using
read_tsv - Select and rename the relevant columns
The resulting object, express, is a list of two tibble items.
# locate the expression text files
expression_files <- c(YY1 = 'data/DataSet_01_18.tsv',
YY2 = 'data/DataSet_01_19.tsv')
# load the expression text files
express <- map(expression_files,
~read_tsv(.x, col_names = FALSE) %>%
dplyr::select(2, 3, 7, 9) %>% #9
setNames(c('tf', 'gene', 'fc', 'pvalue')) %>%
filter(tf %in% c('YY1', 'YY2')) %>%
na.omit())
# show the number of genes with recorded fold-change and p-value
map(express, nrow)
#> $YY1
#> [1] 16595
#>
#> $YY2
#> [1] 16595
# show same genes were recorded for both factors
all(express$YY1$gene %in% express$YY2$gene)
#> [1] TRUE
# show direction and significance of the regulation
table(express$YY1$fc > 0, express$YY1$pvalue < .01)
#>
#> FALSE TRUE
#> FALSE 6785 160
#> TRUE 9280 370
table(express$YY2$fc > 0, express$YY2$pvalue < .01)
#>
#> FALSE TRUE
#> FALSE 7435 132
#> TRUE 8804 224The knockdown of either factor in HeLa cells seem to change the expression of many genes in either directions (Figure 1A&B). Moreover, the changes resulting from the knockdown of the factors individually are correlated (Figure 1C). This suggest that, many of the regulated genes are shared targets of the two factors or they respond similarly to the perturbation of either factor.
# Figure 1
par(mfrow = c(1, 3))
# volcano plot of YY1 knockdown
plot(express$YY1$fc,
-log10(express$YY1$pvalue),
xlab = 'Fold-change (log_2)',
ylab = 'P-value (-log_10)',
xlim = c(-4, 4), ylim = c(0, 6))
title('(A)')
# volcano plot of YY2 knockdown
plot(express$YY2$fc,
-log10(express$YY2$pvalue),
xlab = 'Fold-change (log_2)',
ylab = 'P-value (-log_10)',
xlim = c(-4, 4), ylim = c(0, 6))
title('(B)')
# plot fold-change of YY1 and YY2
plot(express$YY1$fc[order(express$YY1$gene)],
express$YY2$fc[order(express$YY2$gene)],
xlab = 'YY1-knockdown (log_2)',
ylab = 'YY2-knockdown (log_2)',
xlim = c(-4, 4), ylim = c(-4, 4))
title('(C)')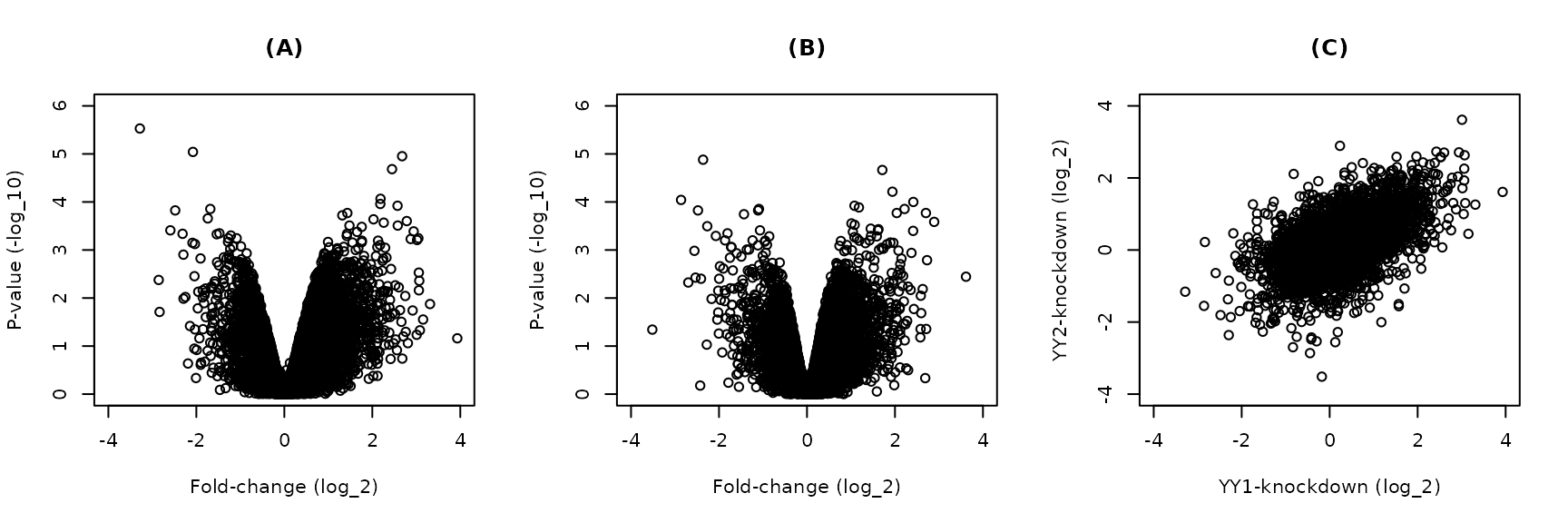
Figure 1. Differential expression between factor knockdown and control HeLa cells. Gene expression was compared between transcription factors knockdown and control HeLa cells. The fold-change and p-values of (A) YY1- and (B) YY2-knockdown are shown as volcano plots. (C) Scatter plot of the fold-change of the YY1- and YY2-knockdown.
Preparing genome annotation
The gene information in express are recorded using the gene SYMBOLS.
- Map SYMBOLS to the ENTREZIDs using
org.Hs.eg.db - Extract the genomic coordinates from
TxDb.Hsapiens.UCSC.hg19.knownGene - Resize the transcripts to 100kb upstream from transcription start sites
# map symbols to entrez ids
symbol_entrez <- AnnotationDbi::select(org.Hs.eg.db,
keys = express$YY1$gene,
columns = 'ENTREZID',
keytype = 'SYMBOL')
# remove unmapped genes
symbol_entrez <- na.omit(symbol_entrez)
# resize regions
genome <- promoters(TxDb.Hsapiens.UCSC.hg19.knownGene,
upstream = 100000,
filter = list(gene_id = symbol_entrez$ENTREZID),
columns = c('tx_id', 'tx_name', 'gene_id'))
# match and add symbols
ind <- match(genome$gene_id@unlistData, symbol_entrez$ENTREZID)
genome$gene <- symbol_entrez$SYMBOL[ind]
# show the first few entries of the mapping data.frame
head(symbol_entrez)
#> SYMBOL ENTREZID
#> 1 IFI44L 10964
#> 2 C7orf57 136288
#> 3 KLHL32 114792
#> 4 TREM2 54209
#> 5 EPHX2 2053
#> 6 LRRC25 126364
# show txdb object
TxDb.Hsapiens.UCSC.hg19.knownGene
#> TxDb object:
#> # Db type: TxDb
#> # Supporting package: GenomicFeatures
#> # Data source: UCSC
#> # Genome: hg19
#> # Organism: Homo sapiens
#> # Taxonomy ID: 9606
#> # UCSC Table: knownGene
#> # Resource URL: http://genome.ucsc.edu/
#> # Type of Gene ID: Entrez Gene ID
#> # Full dataset: yes
#> # miRBase build ID: GRCh37
#> # transcript_nrow: 82960
#> # exon_nrow: 289969
#> # cds_nrow: 237533
#> # Db created by: GenomicFeatures package from Bioconductor
#> # Creation time: 2015-10-07 18:11:28 +0000 (Wed, 07 Oct 2015)
#> # GenomicFeatures version at creation time: 1.21.30
#> # RSQLite version at creation time: 1.0.0
#> # DBSCHEMAVERSION: 1.1
# show the
show(genome)
#> GRanges object with 56521 ranges and 4 metadata columns:
#> seqnames ranges strand | tx_id tx_name
#> <Rle> <IRanges> <Rle> | <integer> <character>
#> uc001abv.1 chr1 760530-860729 + | 21 uc001abv.1
#> uc001abw.1 chr1 761121-861320 + | 22 uc001abw.1
#> uc031pjl.1 chr1 761302-861501 + | 23 uc031pjl.1
#> uc031pjm.1 chr1 761302-861501 + | 24 uc031pjm.1
#> uc031pjn.1 chr1 761302-861501 + | 25 uc031pjn.1
#> ... ... ... ... . ... ...
#> uc011mgb.1 chrUn_gl000223 68588-168787 - | 82934 uc011mgb.1
#> uc011mgc.1 chrUn_gl000223 68588-168787 - | 82935 uc011mgc.1
#> uc011mgd.2 chrUn_gl000223 119531-219730 - | 82936 uc011mgd.2
#> uc031tgg.1 chrUn_gl000223 119531-219730 - | 82937 uc031tgg.1
#> uc031tgh.1 chrUn_gl000223 119531-219730 - | 82938 uc031tgh.1
#> gene_id gene
#> <CharacterList> <character>
#> uc001abv.1 148398 SAMD11
#> uc001abw.1 148398 SAMD11
#> uc031pjl.1 148398 SAMD11
#> uc031pjm.1 148398 SAMD11
#> uc031pjn.1 148398 SAMD11
#> ... ... ...
#> uc011mgb.1 7637 ZNF84
#> uc011mgc.1 7637 ZNF84
#> uc011mgd.2 7574 ZNF26
#> uc031tgg.1 7574 ZNF26
#> uc031tgh.1 7574 ZNF26
#> -------
#> seqinfo: 93 sequences (1 circular) from hg19 genomeNow the two objects can be merged. The merged object, regions, is similarly a GRanges that contains genome and expression information of all common genes.
# make regions by merging the genome and express data
regions <- map(express,
~{
# make a copy of genome
gr <- genome
# match gene names
ind <- match(gr$gene, .x$gene)
# add the expression info to the metadata
mcols(gr) <- cbind(mcols(gr), .x[ind,])
gr
})
# show the first few entries in the GRanges object
regions$YY1
#> GRanges object with 56521 ranges and 8 metadata columns:
#> seqnames ranges strand | tx_id tx_name
#> <Rle> <IRanges> <Rle> | <integer> <character>
#> uc001abv.1 chr1 760530-860729 + | 21 uc001abv.1
#> uc001abw.1 chr1 761121-861320 + | 22 uc001abw.1
#> uc031pjl.1 chr1 761302-861501 + | 23 uc031pjl.1
#> uc031pjm.1 chr1 761302-861501 + | 24 uc031pjm.1
#> uc031pjn.1 chr1 761302-861501 + | 25 uc031pjn.1
#> ... ... ... ... . ... ...
#> uc011mgb.1 chrUn_gl000223 68588-168787 - | 82934 uc011mgb.1
#> uc011mgc.1 chrUn_gl000223 68588-168787 - | 82935 uc011mgc.1
#> uc011mgd.2 chrUn_gl000223 119531-219730 - | 82936 uc011mgd.2
#> uc031tgg.1 chrUn_gl000223 119531-219730 - | 82937 uc031tgg.1
#> uc031tgh.1 chrUn_gl000223 119531-219730 - | 82938 uc031tgh.1
#> gene_id gene tf gene fc
#> <CharacterList> <character> <character> <character> <numeric>
#> uc001abv.1 148398 SAMD11 YY1 SAMD11 -0.54365
#> uc001abw.1 148398 SAMD11 YY1 SAMD11 -0.54365
#> uc031pjl.1 148398 SAMD11 YY1 SAMD11 -0.54365
#> uc031pjm.1 148398 SAMD11 YY1 SAMD11 -0.54365
#> uc031pjn.1 148398 SAMD11 YY1 SAMD11 -0.54365
#> ... ... ... ... ... ...
#> uc011mgb.1 7637 ZNF84 YY1 ZNF84 0.08999
#> uc011mgc.1 7637 ZNF84 YY1 ZNF84 0.08999
#> uc011mgd.2 7574 ZNF26 YY1 ZNF26 -0.47400
#> uc031tgg.1 7574 ZNF26 YY1 ZNF26 -0.47400
#> uc031tgh.1 7574 ZNF26 YY1 ZNF26 -0.47400
#> pvalue
#> <numeric>
#> uc001abv.1 0.05581
#> uc001abw.1 0.05581
#> uc031pjl.1 0.05581
#> uc031pjm.1 0.05581
#> uc031pjn.1 0.05581
#> ... ...
#> uc011mgb.1 0.68511
#> uc011mgc.1 0.68511
#> uc011mgd.2 0.09301
#> uc031tgg.1 0.09301
#> uc031tgh.1 0.09301
#> -------
#> seqinfo: 93 sequences (1 circular) from hg19 genome
# show the names of the columns in the object metadata
names(mcols(regions$YY1))
#> [1] "tx_id" "tx_name" "gene_id" "gene" "tf" "gene" "fc"
#> [8] "pvalue"
# show the width of the regions
unique(width(regions$YY1))
#> [1] 100200Predicting gene targets of individual factors
The standard target analysis includes the identification of associated peaks using associated_peaks and direct targets using direct_targets.
The inputs for these functions are
peaksregions-
regions_col, the column names for regions -
stats_col, the statistics column which is the fold-change in this case.
The resulting objects are GRanges for the identified peaks assigned to the regions or the ranked targets. Several columns is added to the metadata objects of the GRanges to save the calculations.
# get associated peaks
ap <- map2(peaks, regions,
~associated_peaks(peaks=.x,
regions = .y,
regions_col = 'tx_id'))
# show associated_peak return
ap
#> $YY1
#> GRanges object with 160722 ranges and 7 metadata columns:
#> seqnames ranges strand | name
#> <Rle> <IRanges> <Rle> | <character>
#> [1] chr1 762751-762895 * | ID=SRX190209;Name=YY..
#> [2] chr1 762751-762895 * | ID=SRX190209;Name=YY..
#> [3] chr1 762751-762895 * | ID=SRX190209;Name=YY..
#> [4] chr1 762751-762895 * | ID=SRX190209;Name=YY..
#> [5] chr1 762751-762895 * | ID=SRX190209;Name=YY..
#> ... ... ... ... . ...
#> [160718] chrX 154611695-154611857 * | ID=SRX190209;Name=YY..
#> [160719] chrX 154611695-154611857 * | ID=SRX190209;Name=YY..
#> [160720] chrX 154611909-154612034 * | ID=SRX190209;Name=YY..
#> [160721] chrX 154611909-154612034 * | ID=SRX190209;Name=YY..
#> [160722] chrY 21154746-21154873 * | ID=SRX190209;Name=YY..
#> score itemRgb thick assigned_region distance
#> <numeric> <character> <IRanges> <integer> <numeric>
#> [1] 109 #006FFF 762751-762895 21 -47807
#> [2] 109 #006FFF 762751-762895 22 -48397
#> [3] 109 #006FFF 762751-762895 23 -48579
#> [4] 109 #006FFF 762751-762895 24 -48579
#> [5] 109 #006FFF 762751-762895 25 -48579
#> ... ... ... ... ... ...
#> [160718] 388 #00FF72 154611695-154611857 78347 -2110.0
#> [160719] 388 #00FF72 154611695-154611857 78348 -2110.0
#> [160720] 88 #0059FF 154611909-154612034 78347 -1914.5
#> [160721] 88 #0059FF 154611909-154612034 78348 -1914.5
#> [160722] 163 #00A6FF 21154746-21154873 78747 -49796.5
#> peak_score
#> <numeric>
#> [1] 0.0896107
#> [2] 0.0875207
#> [3] 0.0868859
#> [4] 0.0868859
#> [5] 0.0868859
#> ... ...
#> [160718] 0.5574402
#> [160719] 0.5574402
#> [160720] 0.5618165
#> [160721] 0.5618165
#> [160722] 0.0827559
#> -------
#> seqinfo: 56 sequences from an unspecified genome; no seqlengths
#>
#> $YY2
#> GRanges object with 20393 ranges and 7 metadata columns:
#> seqnames ranges strand | name
#> <Rle> <IRanges> <Rle> | <character>
#> [1] chr1 894578-894665 * | ID=SRX1530266;Name=Y..
#> [2] chr1 894578-894665 * | ID=SRX1530266;Name=Y..
#> [3] chr1 894578-894665 * | ID=SRX1530266;Name=Y..
#> [4] chr1 894578-894665 * | ID=SRX1530266;Name=Y..
#> [5] chr1 894578-894665 * | ID=SRX1530266;Name=Y..
#> ... ... ... ... . ...
#> [20389] chrX 154299744-154299864 * | ID=SRX1530266;Name=Y..
#> [20390] chrX 154444690-154444806 * | ID=SRX1530266;Name=Y..
#> [20391] chrX 154444690-154444806 * | ID=SRX1530266;Name=Y..
#> [20392] chrX 154611688-154611827 * | ID=SRX1530266;Name=Y..
#> [20393] chrX 154611688-154611827 * | ID=SRX1530266;Name=Y..
#> score itemRgb thick assigned_region distance
#> <numeric> <character> <IRanges> <integer> <numeric>
#> [1] 150 #0099FF 894578-894665 55 48555.5
#> [2] 150 #0099FF 894578-894665 56 47693.5
#> [3] 150 #0099FF 894578-894665 57 47513.5
#> [4] 150 #0099FF 894578-894665 58 47061.5
#> [5] 150 #0099FF 894578-894665 59 46787.5
#> ... ... ... ... ... ...
#> [20389] 141 #008FFF 154299744-154299864 78345 -49644.0
#> [20390] 125 #007FFF 154444690-154444806 76878 49948.0
#> [20391] 125 #007FFF 154444690-154444806 76879 49948.0
#> [20392] 192 #00C3FF 154611688-154611827 78347 -2128.5
#> [20393] 192 #00C3FF 154611688-154611827 78348 -2128.5
#> peak_score
#> <numeric>
#> [1] 0.0869676
#> [2] 0.0900185
#> [3] 0.0906690
#> [4] 0.0923232
#> [5] 0.0933406
#> ... ...
#> [20389] 0.0832623
#> [20390] 0.0822559
#> [20391] 0.0822559
#> [20392] 0.5570279
#> [20393] 0.5570279
#> -------
#> seqinfo: 41 sequences from an unspecified genome; no seqlengths
# show added columns in the output
names(mcols(peaks$YY1))
#> [1] "name" "score" "itemRgb" "thick"
names(mcols(ap$YY1))
#> [1] "name" "score" "itemRgb" "thick"
#> [5] "assigned_region" "distance" "peak_score"
# show information in the added columns
head(ap$YY1$assigned_region)
#> [1] 21 22 23 24 25 26
head(ap$YY1$distance)
#> [1] -47807 -48397 -48579 -48579 -48579 -48579
head(ap$YY1$peak_score)
#> [1] 0.08961075 0.08752069 0.08688586 0.08688586 0.08688586 0.08688586
# get direct targets
dt <- map2(peaks, regions,
~direct_targets(peaks=.x,
regions = .y,
regions_col = 'tx_id',
stats_col = 'fc'))
# show direct_targets return
dt
#> $YY1
#> GRanges object with 44403 ranges and 13 metadata columns:
#> seqnames ranges strand | tx_id tx_name gene_id
#> <Rle> <IRanges> <Rle> | <integer> <character> <list>
#> 1 chr1 760530-860729 + | 21 uc001abv.1 148398
#> 6 chr1 761121-861320 + | 22 uc001abw.1 148398
#> 11 chr1 761302-861501 + | 23 uc031pjl.1 148398
#> 16 chr1 761302-861501 + | 24 uc031pjm.1 148398
#> 21 chr1 761302-861501 + | 25 uc031pjn.1 148398
#> ... ... ... ... . ... ... ...
#> 160718 chrUn_gl000223 68588-168787 - | 82934 uc011mgb.1 7637
#> 160719 chrUn_gl000223 68588-168787 - | 82935 uc011mgc.1 7637
#> 160720 chrUn_gl000223 119531-219730 - | 82936 uc011mgd.2 7574
#> 160721 chrUn_gl000223 119531-219730 - | 82937 uc031tgg.1 7574
#> 160722 chrUn_gl000223 119531-219730 - | 82938 uc031tgh.1 7574
#> gene tf gene.1 fc pvalue score
#> <character> <character> <character> <numeric> <numeric> <numeric>
#> 1 SAMD11 YY1 SAMD11 -0.54365 0.05581 0.953844
#> 6 SAMD11 YY1 SAMD11 -0.54365 0.05581 0.944972
#> 11 SAMD11 YY1 SAMD11 -0.54365 0.05581 0.942342
#> 16 SAMD11 YY1 SAMD11 -0.54365 0.05581 0.942342
#> 21 SAMD11 YY1 SAMD11 -0.54365 0.05581 0.942342
#> ... ... ... ... ... ... ...
#> 160718 ZNF84 YY1 ZNF84 0.08999 0.68511 0.175405
#> 160719 ZNF84 YY1 ZNF84 0.08999 0.68511 0.175405
#> 160720 ZNF26 YY1 ZNF26 -0.47400 0.09301 0.273345
#> 160721 ZNF26 YY1 ZNF26 -0.47400 0.09301 0.273345
#> 160722 ZNF26 YY1 ZNF26 -0.47400 0.09301 0.273345
#> score_rank stat stat_rank rank
#> <integer> <numeric> <integer> <numeric>
#> 1 14440 -0.54365 7623 0.0558301
#> 6 14621 -0.54365 7623 0.0565299
#> 11 14671 -0.54365 7623 0.0567232
#> 16 14671 -0.54365 7623 0.0567232
#> 21 14671 -0.54365 7623 0.0567232
#> ... ... ... ... ...
#> 160718 37285 0.08999 35329 0.668099
#> 160719 37285 0.08999 35329 0.668099
#> 160720 33771 -0.47400 9775 0.167431
#> 160721 33771 -0.47400 9775 0.167431
#> 160722 33771 -0.47400 9775 0.167431
#> -------
#> seqinfo: 93 sequences from an unspecified genome; no seqlengths
#>
#> $YY2
#> GRanges object with 15296 ranges and 13 metadata columns:
#> seqnames ranges strand | tx_id tx_name gene_id
#> <Rle> <IRanges> <Rle> | <integer> <character> <list>
#> 1 chr1 795967-896166 + | 55 uc001aca.2 339451
#> 2 chr1 796829-897028 + | 56 uc001acb.1 339451
#> 3 chr1 797009-897208 + | 57 uc010nya.1 339451
#> 4 chr1 797461-897660 + | 58 uc001acc.2 339451
#> 5 chr1 797735-897934 + | 59 uc010nyb.1 339451
#> ... ... ... ... . ... ... ...
#> 20388 chrX 154134649-154234848 - | 78342 uc010nvi.1 2157
#> 20389 chrX 154255016-154355215 - | 78343 uc011mzx.1 2157
#> 20391 chrX 154299348-154399547 - | 78345 uc004fmz.2 4515
#> 20392 chrX 154563787-154663986 - | 78347 uc004fnf.3 1193
#> 20393 chrX 154563787-154663986 - | 78348 uc010nvj.1 1193
#> gene tf gene.1 fc pvalue score
#> <character> <character> <character> <numeric> <numeric> <numeric>
#> 1 KLHL17 YY2 KLHL17 -0.39191 0.30656 0.0869676
#> 2 KLHL17 YY2 KLHL17 -0.39191 0.30656 0.0900185
#> 3 KLHL17 YY2 KLHL17 -0.39191 0.30656 0.0906690
#> 4 KLHL17 YY2 KLHL17 -0.39191 0.30656 0.0923232
#> 5 KLHL17 YY2 KLHL17 -0.39191 0.30656 0.0933406
#> ... ... ... ... ... ... ...
#> 20388 F8 YY2 F8 0.32501 0.12452 0.3079124
#> 20389 F8 YY2 F8 0.32501 0.12452 0.5732370
#> 20391 MTCP1 YY2 MTCP1 -0.06521 0.65344 0.0832623
#> 20392 CLIC2 YY2 CLIC2 0.21019 0.35482 0.5570279
#> 20393 CLIC2 YY2 CLIC2 0.21019 0.35482 0.5570279
#> score_rank stat stat_rank rank
#> <integer> <numeric> <integer> <numeric>
#> 1 11139 -0.39191 2527 0.120308
#> 2 11002 -0.39191 2527 0.118829
#> 3 10973 -0.39191 2527 0.118515
#> 4 10902 -0.39191 2527 0.117749
#> 5 10865 -0.39191 2527 0.117349
#> ... ... ... ... ...
#> 20388 5339 0.32501 3699 0.0844090
#> 20389 1707 0.32501 3699 0.0269875
#> 20391 11837 -0.06521 12032 0.6087286
#> 20392 1908 0.21019 6592 0.0537576
#> 20393 1908 0.21019 6592 0.0537576
#> -------
#> seqinfo: 93 sequences from an unspecified genome; no seqlengths
# show added columns in the output
names(mcols(regions$YY1))
#> [1] "tx_id" "tx_name" "gene_id" "gene" "tf" "gene" "fc"
#> [8] "pvalue"
names(mcols(dt$YY1))
#> [1] "tx_id" "tx_name" "gene_id" "gene" "tf"
#> [6] "gene.1" "fc" "pvalue" "score" "score_rank"
#> [11] "stat" "stat_rank" "rank"
# show information in the added columns
head(dt$YY1$score)
#> [1] 0.9538438 0.9449725 0.9423423 0.9423423 0.9423423 0.9423423
head(dt$YY1$score_rank)
#> [1] 14440 14621 14671 14671 14671 14671
head(dt$YY1$rank)
#> [1] 0.05583011 0.05652992 0.05672324 0.05672324 0.05672324 0.05672324To determine the dominant function of a factor,
- We divide the targets into groups based on the effect of the knockdown
- We use the empirical distribution function (ECDF) to show the fraction of targets at a specified regulatory potential value or less.
Because the ranks rather than the absolute value of the regulatory potential is used, the lower the value the higher the potential. Then the groups of targets can be compared to each other or to a theoretical distribution.
# Figure 2
par(mfrow = c(1, 3))
# plot distance by score of associate peaks
plot(ap$YY1$distance, ap$YY1$peak_score,
xlab = 'Distance', ylab = 'Peak Score',
main = '(A)')
points(ap$YY2$distance, ap$YY2$peak_score)
# make labels, colors and groups
labs <- c('Down', 'None', 'Up')
cols <- c('green', 'gray', 'red')
# make three groups by quantiles
groups <- map(dt,~{
cut(.x$stat, breaks = 3, labels = labs)
})
# plot the group functions
pmap(list(dt, groups, c('(B)', '(C)')), function(x, y, z) {
plot_predictions(x$score_rank,
group = y, colors = cols, labels = labs,
xlab = 'Regulatory Potential', ylab = 'ECDF')
title(z)
})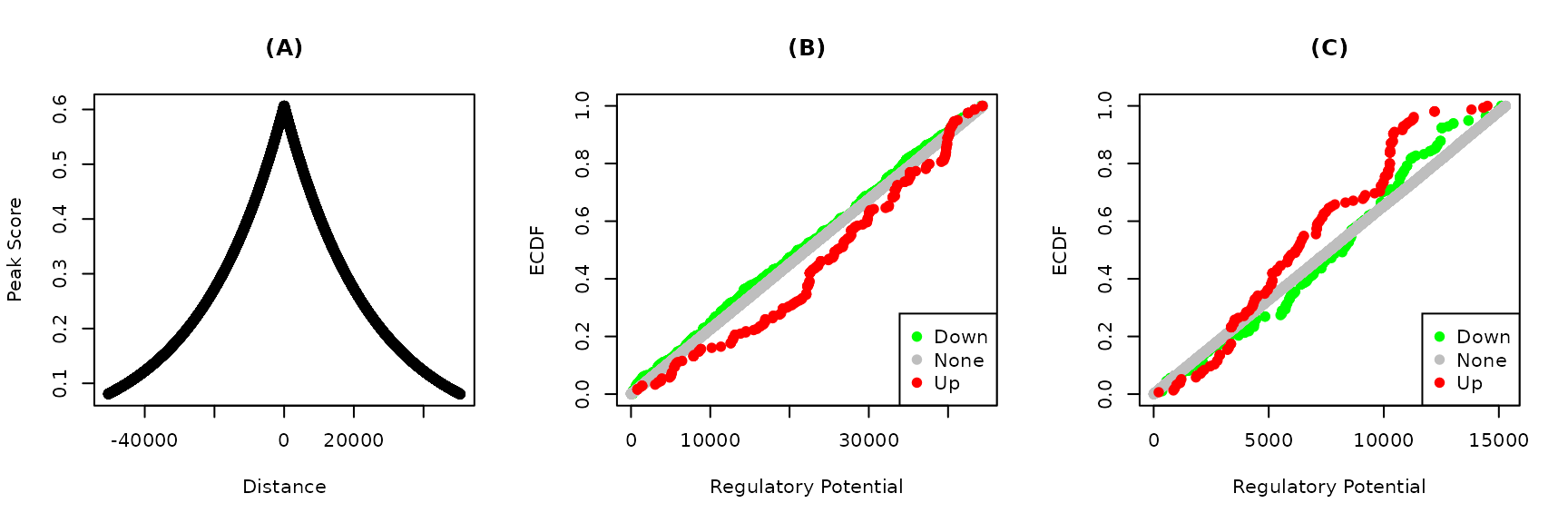
Figure 2. Predicted functions of YY1 and YY2 on their specific targets. Bindings peaks of the transcription factors in HeLa cells were determined using ChIP-Seq. Distances from the transcription start sites and the transformed distances of the (A) YY1 and YY2 peaks are shown. The regulatory potential of each gene was calculated using target. Genes were grouped into up, none or down regulated based on the fold-change. The emperical cumulative distribution functions (ECDF) of the groups of (C) YY1 and (D) YY2 targets are shown at each regulatory potential value.
The scores of the individual peaks are a decreasing function of the distance from the transcription start sites.
The closer the factor binding site from the start site the lower the score. The distribution of these scores is very similar for both factors (Figure 2A). The ECDF of the down-regulated targets of YY1 is higher than that of up- and none-regulated targets (Figure 2B). Therefore, the absence of YY1 on its targets result in aggregate in their down regulation.
The opposite is true for YY2 where more high ranking targets are up-regulated by the factor knockdown (Figure 2C).
# Table 2
# test individual factor functions
map2(dt, groups,
~test_predictions(.x$rank,
group = .y,
compare = c('Down', 'Up')))
#> $YY1
#>
#> Two-sample Kolmogorov-Smirnov test
#>
#> data: x and y
#> D = 0.78597, p-value < 2.2e-16
#> alternative hypothesis: two-sided
#>
#>
#> $YY2
#>
#> Two-sample Kolmogorov-Smirnov test
#>
#> data: x and y
#> D = 0.40354, p-value = 1.075e-12
#> alternative hypothesis: two-sidedTo formally test these observations, we use the Kolmogorov-Smirnov (KS) test. The distribution of the two groups are compared for equality. If one lies one either side of the other then they must be drawn from different distributions.
Here, we compared the up- and down-regulated functions for both factors (Table 2).
In both cases, the distribution of the two groups were significantly different from one another.
Predicting the shared targets of the two factors
Similar to the previous analysis,
- Identify shared/common peaks using
subsetByOverlaps. - Pass two columns to
stats_colto use the fold-changes from both factors -
common_peaksandboth_regionsare the main inputs
# merge and name peaks
common_peaks <- GenomicRanges::reduce(subsetByOverlaps(peaks$YY1, peaks$YY2))
common_peaks$name <- paste0('common_peak_', 1:length(common_peaks))
# bind express tables into one
both_express <- bind_rows(express) %>%
pivot_wider(names_from = tf,
values_from = c(fc, pvalue))
# match symbols between genome and expression objects
ind <- match(genome$gene, both_express$gene)
# subset genome by genes with expression info
both_regions <- genome[genome$gene %in% both_express$gene]
# add expression info to gene info
mcols(both_regions) <- cbind(mcols(both_regions), both_express[ind,])
# get associated peaks with both factors
common_ap <- associated_peaks(peaks = common_peaks,
regions = both_regions,
regions_col = 'tx_id')
# get direct targets of both factors
common_dt <- direct_targets(peaks = common_peaks,
regions = both_regions,
regions_col = 'tx_id',
stats_col = c('fc_YY1', 'fc_YY2'))-
associated_peaksis the same as before. -
direct_targetsis the same but thestatand thestat_rankcarry the product of the two statistics provided in the previous step and the rank of that product.
The output can also be visualized the same way. The targets are divided into three groups based on the statistics product.
- When the two statistics agree in the sign, the product is positive. This means the knockdown of either transcription factor results in same direction change in the target gene expression. Therefore, the two factors would cooperate if they bind to the same site on that gene.
- The reverse is true for targets with opposite signed statistics. On these targets, the two factors would be expected to compete for inducing opposing changes in the expression.
# Figure 3
par(mfrow = c(1, 2))
# plot distiace by score for associated peaks
plot(common_ap$distance,
common_ap$peak_score,
xlab = 'Distance',
ylab = 'Peak Score')
title('(A)')
# make labels, colors and gorups
labs <- c('Competitive', 'None', 'Cooperative')
cols <- c('green', 'gray', 'red')
# make three groups by quantiles
common_groups <- cut(common_dt$stat,
breaks = 3,
labels = labs)
# plot predicted function
plot_predictions(common_dt$score_rank,
group = common_groups,
colors = cols, labels = labs,
xlab = 'Regulatory Interaction', ylab = 'ECDF')
title('(B)')
Figure 3. Predicted function of YY1 and YY2 on their shared targets. Shared bindings sites of YY1 and YY2 in HeLa cells were determined using the overlap of the individual factor ChIP-Seq peaks. (A) Distances from the transcription start sites and the transformed distances of the shared peaks are shown. The regulatory interaction of each gene was calculated using target. Genes were grouped into cooperatively, none or competitively regulated based on the the product of the fold-changes from YY1- and YY2-knockdown. (B) The emperical cumulative distribution functions (ECDF) of the groups of targets are shown at each regulatory potential value.
- The common peaks distances and scores take the same shape (Figure 3A).
- The two factors seem to cooperate on more of the common target than any of the two other possibilities (Figure 3B).
This observation can be tested using the KS test. The curve of the cooperative targets lies above that of none and competitively regulated targets (Table 3).
# Table 3
# test factors are cooperative
test_predictions(common_dt$score_rank,
group = common_groups,
compare = c('Cooperative', 'None'),
alternative = 'greater')
#>
#> Two-sample Kolmogorov-Smirnov test
#>
#> data: x and y
#> D^+ = 0.30253, p-value = 0.01034
#> alternative hypothesis: the CDF of x lies above that of y
# test factors are more cooperative than competitive
test_predictions(common_dt$score_rank,
group = common_groups,
compare = c('Cooperative', 'Competitive'),
alternative = 'greater')
#>
#> Two-sample Kolmogorov-Smirnov test
#>
#> data: x and y
#> D^+ = 0.29706, p-value = 0.01263
#> alternative hypothesis: the CDF of x lies above that of ySummary
- I presented a workflow for predicting the direct targets of a transcription factor by integrating binding and expression data.
- The
targetpackage implements the BETA algorithm to rank targets based on the distance of the ChIP peaks of the transcription factor in the genes and the differential expression from the factor perturbation. - To predict the combined function of two factors, two sets of data are used to find the shared peaks and the product of their differential expression.
References
Chen, Li, Toshi Shioda, Kathryn R. Coser, Mary C. Lynch, Chuanwei Yang, and Emmett V. Schmidt. 2010. “Genome-wide analysis of YY2 versus YY1 target genes.” Nucleic Acids Research 38 (12): 4011–26. https://doi.org/10.1093/nar/gkq112.
Michaud, Joëlle, Viviane Praz, Nicole James Faresse, Courtney K Jnbaptiste, Shweta Tyagi, Frédéric Schütz, and Winship Herr. 2013. “HCFC1 is a common component of active human CpG-island promoters and coincides with ZNF143, THAP11, YY1, and GABP transcription factor occupancy.” Genome Research 23 (6): 907–16. https://doi.org/10.1101/gr.150078.112.
Wu, Xiao-Nan, Tao-Tao Shi, Yao-Hui He, Fei-Fei Wang, Rui Sang, Jian-Cheng Ding, Wen-Juan Zhang, et al. 2017. “Methylation of transcription factor YY2 regulates its transcriptional activity and cell proliferation.” Cell Discovery 3: 17035. https://doi.org/10.1038/celldisc.2017.35.